
Reason 9 download Archives

reason 9 download Archives
PRIDE Archive
Frequently Asked Questions - FAQ
The FAQ covers the most frequent problems encountered during the PRIDE submission and publication process and their resolutions. In case you have a particular issue in mind please try to search the page with a keyword by using Ctrl+F (Windows, Linux) or ⌘-F (Mac) to find the relevant item. Alternatively you can browse through the table of content on the top of the page from where the particular FAQ items are linked. If you cannot find the answer to your question in this FAQ or other help pages then please contact us at pride-support@ebi.ac.uk for advice.
The FAQ is divided into 9 sections:
1. Submission options - What submission options do I have?
2. Submission process - How to do a submission?
3. Data access policy - How can the data be accessed?
4. Post-submission process - How do I modify, reference and make datasets public?
5. Quality Control - How can I annotate my data better?
6. Recommendations for popular proteomics software tools and search engines - What fits my custom pipeline?
7. Recommendations for specific proteomics experiment types - What might fit my bleeding edge research?
8. Related journal guidelines - What are the known requirements of different proteomics journals?
9. Troubleshooting
1. Submission options - What submission options do I have?
What is ProteomeXchange?
What are the PX Submission options via PRIDE?
What is a Complete ProteomeXchange Submission?
What is a Partial ProteomeXchange Submission?
2. Submission process - How to do a submission?
Register
How to manually set the proxy details with the PX Submission Tool?
What type of machine raw files are supported for a PX Submission?
Can PRIDE XML files without identifications be submitted for a Complete PX Submission?
Can mzIdentML files referencing raw files be submitted for a Complete PX Submission?
Can mzIdentML result files and PRIDE XML result files together be submitted for a Complete PX Submission?
Generating MzIdentML files for a Complete Submission with a workflow of different tools, what to be aware of?
How many PX identifiers can be issued per study/manuscript, can a dataset be split into further partitions?
How to export the summary file with the PX Submission Tool?
How to generate the PX Submission Summary File without the PX Submission Tool?
3. Data access policy - How can the data be accessed?
Are the uploaded datasets kept private by default upon a ProteomeXchange Submission?
How to access private datasets?
4. Post-submission process - How do I modify, reference and make datasets public?
How can I modify the original dataset?
How to reference a PX accession in a manuscript?
How to release a dataset publicly?
5. Quality Control - How can I annotate my data better?
What are the delta m/z values displayed in PRIDE Inspector Peptide View?
What to do when you see delta m/z outliers in PRIDE Inspector?
6. Recommendations for popular proteomics software tools and search engines - What fits my custom pipeline?
What kind of Scaffold files are recommended for a PX Submission?
What kind of MaxQuant files are recommended for a PX Submission?
What kind of Proteome Discoverer files are recommended for a PX Submission?
What kind of ProteinPilot files are recommended for a PX Submission?
7. Recommendations for specific proteomics experiment types - What might fit my bleeding edge research?
I have mass spectrometry imaging data, what should I do?
I have multi-omics data uploaded to different databases, what should I do?
How to handle newly discovered, unreported modifications?
8. Related journal guidelines - What are the known requirements of different proteomics journals?
Does my PX dataset fulfil the requirements of the journal Molecular and Cellular Proteomics?
Does my PX dataset fulfil the requirements of the journal Proteomics?
9. Troubleshooting
PX Submission Tool does not launch
PX Submission tool launches, but I cannot login / make a submission
How to switch the PX Submission Tool from Aspera to FTP?
PRIDE Converter 2 Tool does not launch or cannot convert my files
PRIDE Converter 2 Tool still has problems converting my files
How do I update my PRIDE account information?
PRIDE Inspector Java Web Start does not launch
PRIDE Inspector Tool does not launch
Aspera does not connect
Aspera connects, but transfers 0% of my files
I cannot to log in with the PX Submission tool
All my Java applications don't launch! I'm on a Mac...
1. Submission options - What submission options do I have?
What is ProteomeXchange?
The ProteomeXchange consortium has been set up to provide a single point of submission of MS proteomics data to the main existing proteomics repositories, and to encourage the data exchange between them for optimal data dissemination.
Current members accepting submissions are:
-The PRIDE PRoteomics IDEntifications database at the European Bioinformatics Institute focusing mainly on shotgun mass spectrometry proteomics data. However, PRIDE can store data coming from other data workflows as well.
- MassIVE at the University of California (San Diego), also focusing mainly on shotgun mass spectrometry proteomics data.
- PeptideAtlas/PASSEL focusing on SRM/MRM datasets.
What are the PX Submission options via PRIDE?
The default way of submitting data to PRIDE is following the ProteomeXchange (PX) consortium (www.proteomexchange.org) guidelines. This paper in PROTEOMICS (open access) explains in detail the process.
The figure below summarizes the overall submission process submitters will have to follow up to the point of uploading their datasets:

Each submitted PX dataset will contain:
- peptide/protein identification files (called ‘RESULT’),
- mass spectrometer output files (called ‘RAW’), which are either machine raw files or not heavily processed files in a XML-based format such as mzXML or mzML,
- other files like peak list files (called ‘PEAK’), search engine output files (called ‘SEARCH’), quantification files and different post-processing files, amongst others.
The current version of pipeline does not explicitly support quantification results. However, quantification result files can be submitted as accompanying (‘QUANT’) files.
There are two different submission workflows depending on whether peptide/protein identification results can be submitted in a format that can be handled by PRIDE (mzIdentML, PRIDE XML) or not. Otherwise, a "Partial Submission" can be done.
What is a Complete ProteomeXchange Submission?
Two different submission types are available: ‘complete’ and ‘partial’. In both cases ‘RAW’ files and metadata are mandatory. Also in both cases processed identification results are also required, but the difference has to do with the file format in which these results are provided. For complete submissions, processed identification results are provided as either mzIdentML or PRIDE XML files. The recommended submission subtype is a Complete Submission, but alternatively Partial Submissions are accepted as well.
A complete submission ensures that the processed results data can be integrated in the PRIDE database and that the connection between the PSMs and the mass spectra can be directly established. This connection makes the peptide/protein identification data searchable in the web interface, and also means that the data can be visualised using PRIDE Inspector.
3A. Complete Submission: mzIdentML or PRIDE XML-based
The 2 subtypes of Complete Submissions are either mzIdentML- or PRIDE XML-based. Complete Submissions mixing the 2 types of ‘RESULT’ files are not allowed.
An mzIdentML-based Complete Submission requires 3 types of files:
- "Result" files: mzIdentML 1.1 files with identifications provided. In the PX Submission tool they should be tagged as “RESULT” files. It is also highly recommended to check your mzIdentML files before submission using PRIDE Inspector. Older mzIdentML version 1.0 files are not supported.
- "Peak list" files: Since the mzIdentML files themselves do not contain the spectra information it is mandatory to provide the peak list files (eg. mgf files) that were used for the original search and are referenced in the mzIdentML file. These are different from the provided mandatory "RAW" files. In the submission tool they should be tagged as “PEAK” and the submission tool will try to automatically map the peak files to the mzIdentML file where they are listed.
- "Raw" files: the MS instrument output files, for instance Thermo RAW files. As an alternative, lightly processed mzML, mzXML or mzData files are acceptable if MS1 level spectra information is available and the different peak processing steps are known. In the submission tool they should be tagged as “RAW”.
Please check our Guide to generate mzIdentML files. It is possible that you are already using a pipeline/search engine where mzIdentML files are amongst the native search engine output formats. mzIdentML files can be created/exported already with numerous tools, please see a list here.
Besides the three mandatory file types above, there are optional and recommended file types that can be prepared and uploaded as well:
- Search engine output files: The original output from your search engine or your analysis pipeline used by you for further post-processing, such as Mascot .dat files, Trans Proteomics Pipeline (TPP) pep.xml and/or prot.xml files among many others. In case your search engine generated mzIdentML files you already provided them as "Result" files. They search engine files should contain peptide/protein identification results. In the submission tool they should be tagged as “SEARCH”.
- Quantification result files: In many cases current mass spectometry proteomics studies do involve a quantitative analysis on the peptides/proteins present in the samples. Quantification related files reporting on peptide/protein quantitative values/ratios can be provided and tagged as "QUANT" in the submission tool. In the future we plan to support the PSI standard format mzTab, to handle quantitative submissions.
- Sequence database files: Sequence database file (usually in FASTA format) that was used to perform the mass spectral search. If you have used custom search databases you can provide those as well. Sequence database files (both protein and DNA) are labelled as "FASTA" in the tool.
- Spectrum libraries: Spectral library file that was used for performing the mass spectrometry search. In the PX Submission Tool they should be tagged as "SPECTRUM_LIBRARY".
- Gel image files: In case two-dimensional gel electrophoresis has been used as a separation method the gel image files can be provided. In the PX Submission Tool they should be tagged as "GEL".
- Other files: Everything else that did not fit into the categories above for instance protein inference files generated by post-processing of the search engine results or R scripts used for data analysis. In the submission tool they should be tagged as ‘OTHER’.
A PRIDE XML-basedComplete Submission requires 2 types of files:
- Result files: fully supported by PRIDE: PRIDE XML files with protein/peptide identifications provided. In the PX Submission Tool they should be tagged as “RESULT”. It is also recommended to check your PRIDE XML files before submission using PRIDE Inspector.
- Raw files: the MS instrument output files, for instance Thermo RAW files. As an alternative, lightly processed mzML, mzXML or mzData files are acceptable if MS1 level spectra information is available and the different peak processing steps are known. In the submission tool they should be tagged as “RAW” and mapped to the corresponding "RESULT" files.
Try to create PRIDE XML files using the PRIDE Converter 2 tool. Please take a moment to review our Guide to generate PRIDE XML files concerning the input files you can use for PRIDE XML generation. There are other tools that can produce PRIDE XML files, not mantained by the PRIDE team, like PeptideShaker, Waters PLGS, Proteios , EasyProt, MIAPE Extractor (ProteoRed), or the original PRIDE Converter tool (no longer further developed or maintained).
Besides the two mandatory file types above, there are optional and recommended file types that can be prepared and uploaded as well:
- "Peak list" files. It is strongly recommended to provide the peak list files (eg. mgf files) that were used for the original search and these are different from the provided mandatory raw files. In the submission tool they should be tagged as “PEAK”.
- Search engine output files: the original output from your search engine or your analysis pipeline, such as Mascot .dat files, Trans Proteomics Pipeline (TPP) pep.xml and/or prot.xml files or mzIdentML files, among many others. They should contain the peptide/protein identifications. In the submission tool they should be tagged as “SEARCH”.
- Quantification result files: In many cases current mass spectometry proteomics studies do involve a quantitative analysis on the peptides/proteins present in the samples. Quantification related files reporting on peptide/protein quantitative values/ratios can be provided and tagged as "QUANT" in the submission tool. In the future we plan to support the PSI standard format mzTab, to handle quantitative submissions.
- Sequence database files: Sequence database file (usually in FASTA format) that was used to perform the mass spectral search. If you have used custom search databases you can provide those as well. Sequence database files (both protein and DNA) are labelled as "FASTA" in the tool.
- Spectrum libraries: Spectral library file that was used for performing the mass spectrometry search. In the PX Submission Tool they should be tagged as "SPECTRUM_LIBRARY".
- Gel image files: In case two-dimensional gel electrophoresis has been used as a separation method the gel image files can be provided. In the PX Submission Tool they should be tagged as "GEL".
- Other files: Everything else that did not fit into the categories above for instance protein inference files generated by post-processing of the search engine results or R scripts used for data analysis. In the submission tool they should be tagged as ‘OTHER’.
In case of a Complete Submission a DOI (Digital Object Identifier) will be assigned to your dataset and its traceability will then be higher. That is good for your data and good for the community.
What is a Partial ProteomeXchange Submission?
In this case processed identification results are provided in other formats (search engine output files, ‘SEARCH’ files). The processed identification results cannot be integrated and made searchable in PRIDE (peptide and protein identifications), or visualized using PRIDE Inspector. However, all the files are available to download via FTP or Aspera. This mechanism allows data generated from software that cannot export to standard formats, or from other experimental approaches (e.g. top down proteomics, Data independent adquisition, etc) to be deposited into PRIDE.
You should only choose this option by default if your search results cannot be converted/exported to PRIDE XML or mzIdentML v1.1 (plus the accompanying spectra). On the other hand in case the generated 'RESULT' files for a Complete Submission do not pass the PRIDE validation pipeline and fixing the problems is not guaranteed within an acceptable timeframe a Partial Submission is also possible.
‘RAW’ files need to be provided together with search engine output files (‘SEARCH’). Uploading peak list (‘PEAK’), quantification and other types of files (‘QUANT’, ‘GEL’, 'FASTA', 'SPECTRUM_LIBRARY' or ‘OTHER’) is possible but not enforced. As a result, you will be issued with a ProteomeXchange accession number but not with a DOI. Once it is made public, your dataset will be available to download via FTP.
A Partial Submission requires 2 types of files:
- Search engine result files: (called ‘SEARCH’): the original output files from your search engine or your analysis pipeline, Trans-Proteomic Pipeline (TPP) pep.xml and/or prot.xml files, or MaxQuant text output files, among many others. They should contain the peptide/protein identifications. In the submission tool they should be tagged as ‘SEARCH’.
- Raw files (called ‘RAW’): MS instrument binary output files, such as Thermo RAW files, BRUKER .baf files or not heavily processed mzXML or mzML files. If your ‘RAW’ files are organized in directories instead of individual files, please compress them into one individual file (for instance to .zip) before upload. In the submission tool they should be tagged as ‘RAW’.
Besides the two mandatory file types above, there are optional and recommended file types that can be prepared and uploaded as well:
- "Peak list" files. It is strongly recommended to provide the peak list files (eg. mgf files) that were used for the original search and these are different from the provided mandatory raw files. In the submission tool they should be tagged as “PEAK”.
- Search engine output files: the original output from your search engine or your analysis pipeline, such as Mascot .dat files, Trans Proteomics Pipeline (TPP) pep.xml and/or prot.xml files or mzIdentML files, among many others. They should contain the peptide/protein identifications. In the submission tool they should be tagged as “SEARCH”.
- Quantification result files: In many cases current mass spectometry proteomics studies do involve a quantitative analysis on the peptides/proteins present in the samples. Quantification related files reporting on peptide/protein quantitative values/ratios can be provided and tagged as "QUANT" in the submission tool. In the future we plan to support the PSI standard format mzTab, to handle quantitative submissions.
- Sequence database files: Sequence database file (usually in FASTA format) that was used to perform the mass spectral search. If you have used custom search databases you can provide those as well. Sequence database files (both protein and DNA) are labelled as "FASTA" in the tool.
- Spectrum libraries: Spectral library file that was used for performing the mass spectrometry search. In the PX Submission Tool they should be tagged as "SPECTRUM_LIBRARY".
- Gel image files: In case two-dimensional gel electrophoresis has been used as a separation method the gel image files can be provided. In the PX Submission Tool they should be tagged as "GEL".
- Other files: Everything else that did not fit into the categories above for instance protein inference files generated by post-processing of the search engine results or R scripts used for data analysis. In the submission tool they should be tagged as ‘OTHER’.
To perform a partial Submission means that a PX accession number will be assigned to your files but PRIDE assay accession numbers won't be issued. Also, you won't have a DOI (Digital Object Identifier) assigned to your dataset.
2. Submission process - How to do a data submission?
Register
If you do not already have a PRIDE account, create one here. Currently we don't send out automatic emails upon succesful registration. Please contact pride-support@ebi.ac.uk if your login information is not valid after 24 hours following registration.
What type of machine raw files are supported for a PX Submission?
Thermo .RAW, ABSCIEX .wiff, .wiff.scan, Agilent .d/, Waters .raw/ • imzML, Shimadzu .run/, Bruker .yep, Bruker .baf
As an alternative, lightly processed mzML, mzXML or mzData files are acceptable if MS1 level spectra information is available and the different peak processing steps are known.
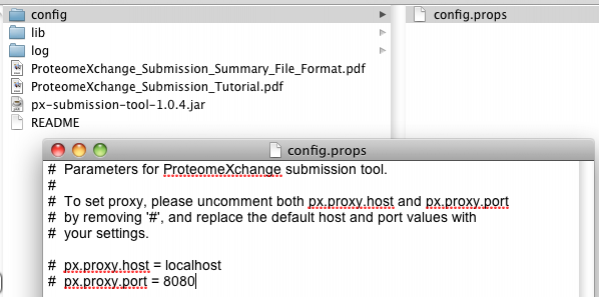
How to manually set the proxy details with the PX Submission Tool?
From version 1.0.4 and up the proxy details can be manually set with the PX Submission Tool. The tool's working directory contains a 'config' folder with a text file 'config.props' There the proxy host and port can be manually set by overwriting and uncommenting the proxy details below:
# px.proxy.host = localhost
# px.proxy.port = 8080
See also the screenshot:
Can PRIDE XML files without identifications be submitted for a Complete PX Submission?
No. PRIDE XML files without protein/peptide identifications do not qualify for a Complete PX Submission. You will have to investigate the options to provide identifications as well. Complete submissions enable the direct connection between the reported identifications and the mass spectra. This is not possible if identifications are not included in the submitted PRIDE XML files.
In case you are unable to produce PRIDE XML files with identifications included you can still do a Partial PX Submission. You can provide your identification files as 'SEARCH' files and map 'RAW' files for that.
Can mzIdentML files referencing raw files be submitted for a Complete PX Submission?
No. Although mzIdentML files referencing raw files can be generated and are valid files, they cannot be used for Complete submissions. Complete submissions enable the direct connection between the reported identifications and the mass spectra. This is not possible for us if raw files are referenced.
MzIdentML files should reference the following peak list/spectra files: mgf, pkl, ms2, dta, apl, mzXML, mzML and mzData.
Can mzIdentML result files and PRIDE XML result files together be submitted for a Complete PX Submission?
No. In order to avoid the heterogeneous representation of 'RESULT' files in mixed formats it is not allowed within the same dataset to provide the results as mzIdentML and as PRIDE XML files at the same time.
Generating mzIdentML files for a Complete Submission with a workflow of different tools, what to be aware of?
It is not uncommon in current analysis workflows to chain together different tools for downstream processing. For instance a default database search is done with Mascot and then TPP was used to do the post-processing of the results. In case different analysis tools are used to generate the final reported results for a Complete Submission the most important issue to be aware of is that the mzIdentML file generated at the end containing the final reported results must still be able to link correctly to the peak list file (eg. mgf file) that was originally searched. These peak list lifes (the ones referenced in the mzIdentML files) need be submitted for qualifying for a Complete Submission. If the link is lost, the PSMs linking the spectra to peptide identifications cannot be recovered and a Partial Submission is then the recommended way to upload data to ProteomeXchange, please see http://www.ebi.ac.uk/pride/help/archive/faq#partial-PX.
How many PX identifiers can be issued per study/manuscript, can a dataset be split into further partitions?
Sometimes submitters plan to split the dataset belonging to the same study/manuscript/project into many different dataset partitions due to different reasons. In this case it is a recommended practice to contact the PRIDE Curator Team viapride-support@ebi.ac.uk in order to agree on the structure of the dataset splits before actual data upload, desirably.
By default we issue one identifier per project unless there's a strong reason to split data like samples from different species have been used, or different instrumentation has been applied. Even in case of a justified way of splitting a dataset into partitions there should be no more than 5 identifiers issued by default. If the curators know the dataset structure in advance they will make an effort to assign a continuous range of PX Identifiers to the dataset splits. However there is no guarantee that this could be achieved since ProteomeXchange has a shared identifier space and other PX partner repositories can issue identifiers too.
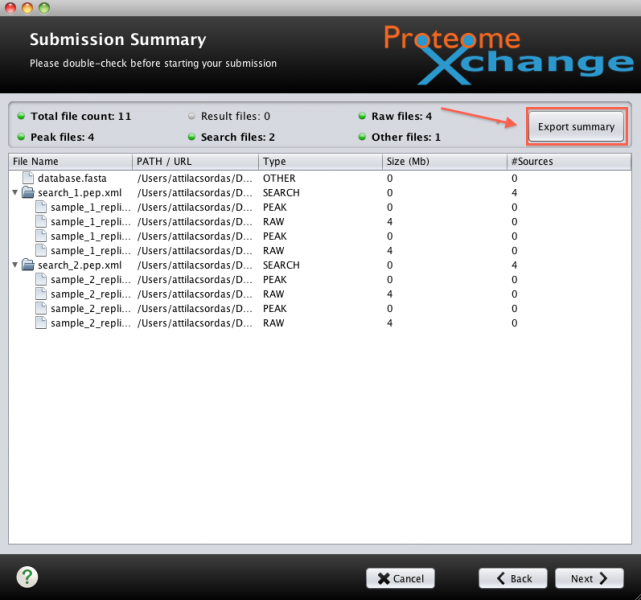
How to export the summary file with the PX Submission Tool?
For some bulk submissions or for direct Aspera upload the summary.px file can still be generated and exported with the PX Submission Tool, although the files won't actually be uploaded with the tool itself.
When using the PX tool there is an "Export Summary" button that you can click after you have done the mappings and that will export the summary.px. It is necessary to have all the files available in a folder so this folder will be used to supply all the file names. But the data won't be actually uploaded, only the summary file exported. See screenshot.
You don't have to start the actual file upload that way. You can send that small file to the database curators via e-mail and upload the files via Aspera.
How to generate the PX Submission Summary File without the PX Submission Tool?
In case of bulk submissions (e.g. if Aspera is used) and there are too many files to handle them with the PX Submission Tool, the px summary files can be generated by scripting. Details of the tab delimited PX Submission format can be found in the ProteomeXchange_Submission_Summary_File_Format.pdf that is distributed with the PX Submission Tool and can be downloaded from here. Example submission summary files (Complete and Partial) can be downloaded form here.
3. Data access policy - How can the data be accessed?
Are the uploaded datasets kept private by default upon a ProteomeXchange Submission?
Yes. All submissions to ProteomeXchange via PRIDE are private by default and the curators only make a particulat dataset public once it is requested by the submitter or we find out that the paper referencing the dataset has been published.
How to access private datasets?
Submitted datasets are 'private' by default, which means you need to be logged-in to view your data. We however also create a reviewer account for your submission (with an username and password) which you can include in your manuscript. The PX reviewer account will give you access to all of the files belonging to your submission. You can access the private dataset files in two ways: via the PRIDE Archive web page or via PRIDE Inspector.
PRIDE Archive web page
The PRIDE Archive web site is available at http://www.ebi.ac.uk/pride/archive. Registered submitters can use their personal accounts or the reviewer accounts to access and download the individual PX datasets. For every submission there is a separate reviewer account generated.
Please navigate first to the login page available at http://www.ebi.ac.uk/pride/archive/login (see Figure):

PRIDE Inspector
For this option you need to use PRIDE Inspector.
The following applies for both Complete and Partial Submissions:
Open PRIDE Inspector by clicking on the pride-inspector-<version-number>.jar file in the tool's working directory -> Private Download -> ProteomeXchange -> PX reviewer account details. You can open the PRIDE XML and mzIdentML 'RESULT' files with PRIDE Inspector or just download all the files that you wish to investigate.

Downloading data with the reviewer account using PRIDE Inspector' Private Download option.
4. Post-submission process - How do I modify, reference and make datasets public?
How can I modify the original dataset?
i., if you need to modify metadata only, for example free text based descriptions (title, project description, sample and data processing protocols), or cv parameters like instrument, species information, please contact PRIDE supoort via emal at pride-support@ebi.ac.uk and we will make the changes for you.
The other 3 options do apply in case you need to modifiy the data files: modify or delete existing files or upload new ones.
ii., In case you have used the PX Submission Tool and you need to add additional 'RAW' files and accompanying 'RESULT' or 'SEARCH' files, you need to resubmit the whole dataset again. Please click the 'resubmisison' option on the first window of the PX Submission Tool and supply the issued PX identifier. The PRIDE Curator Team then will resubmit the dataset and keep the originally issued identifier.
iii., In case you need to add to a small number of additional 'OTHER' (like csv, plain text files, spreadsheets, scripts) we can provide you with FTP details to upload and can add these to the original dataset without you resubmitting the whole dataset.
iv., In case of an Aspera bulk submission you have to upload the modified and/or missing files into a new subdirectory within your target directory and regenerate the px submission summary file to reference and mapp all the old, the modified and the new files again.
How to reference a PX accession number in a manuscript?
By default we recommend to add the following sentence to your manuscript (typically in "Material and Methods" or just before/in the "Acknowledgements"):
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [1] with the dataset identifier <PXD000xxx>."
In case of Complete PX Submissions an extra DOI can be added to this:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium [1] via the PRIDE partner repository with the dataset identifier <PXD000xxx> and DOI 10.6019/<PXD000xxx>."
[1] Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA,Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolomé S, Apweiler R,Omenn GS, Martens L, Jones AR, Hermjakob H (2014). ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nature Biotechnol. 30(3):223-226. PubMed PMID:24727771.
Additionally we'd like to ask you to also put this information in a much abridged form into the abstract itself, like this: "The data have been deposited to the ProteomeXchange with identifier <PXD000xxx>." See for example this Chromosome-Centric Human Proteome Project dataset and paper: http://www.ncbi.nlm.nih.gov/pubmed/?term=23312004 and other examples on PubMed. A PX Identifier in the abstract makes the dataset much more visible and accessible.
Otherwise, for general PRIDE reference, please use:
Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, O'Kelly G, Schoenegger A, Ovelleiro D, Perez-Riverol Y, Reisinger F, Rios D, Wang R, Hermjakob H. The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013 Jan 1;41(D1):D1063-9. doi: 10.1093/nar/gks1262. Epub 2012 Nov 29. PubMed PMID:23203882.
If you have used the PRIDE Inspector tool, please cite: Wang R, Fabregat A, Rios D, Ovelleiro D, Foster JM, Cote RG, Griss J, Csordas A, Perez-Riverol Y, Reisinger F, Hermjakob H, Martens L, Vizcaino JA (2012). PRIDE Inspector: a tool to visualize and validate MS proteomics data.Nat Biotechnol. 30(2):135-7. PubMed PMID: 22318026. If you have used the PRIDE Converter 2 tool, please cite: Côté RG, Griss J, Dianes JA, Wang R, Wright JC, van den Toorn HWP, van Breukelen B, Heck AJR, Hulstaert N, Martens L, Reisinger F, Csordas A, Ovelleiro D, Perez-Riverol Y, Barsnes H, Hermjakob H, Vizcaíno JA (2012). The Proteomics IDEntifications (PRIDE) Converter 2 framework: an improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Mol Cell Proteomics 11: 1682-1689. PubMed PMID: 22949509. |
How to release a dataset publicly?
By default, your data will be made publicly available after your manuscript has been accepted, or when we have your instructions to do so. While we may also receive acceptance notifications from some journals (e.g. Proteomics, Wiley), we would like to ask all submitters to kindly notify us separately. Otherwise, it can happen that we don’t now that your manuscript is already published. You can notify us two ways:
A), Via the new PRIDE Archive web site (http://www.ebi.ac.uk/pride/archive). Once you have logged in with your user account at http://www.ebi.ac.uk/pride/archive/login you can click the green “Publish” buttons located next to your unpublished datasets. Here you can provide details for your dataset and submit a web form, please see Figure.

5. Quality Control - How can I annotate my data better?
What are the delta m/z values displayed in PRIDE Inspector Peptide View?
PRIDE Inspector calculates the ‘delta m/z’ values for the reported identified peptides by calculating the difference between the experimentally detected m/z value (corresponding to the precursor peptide ion m/z) and the theoretical mass of the peptide identified. If the resulting value is outside of a normal range (depending on the accuracy of the mass spectrometer used), this constitutes a good indication that something has gone wrong while annotating the data. For instance, an outlier value can indicate whether the precursor charge was wrongly assigned, or the protein modifications were not reported correctly. In the PRIDE Inspector ‘Peptide View’, the delta m/z values are displayed. Currently, the delta m/z values outside of the −4.0 to +4.0 m/z units range are highlighted in red, while the normal values are highlighted in green. See details in this article.

What to do when you see delta m/z outliers in PRIDE Inspector?
PRIDE Inspector calculates the ‘delta m/z’ values for the reported identified peptides by calculating the difference between the experimentally detected m/z value (corresponding to the precursor peptide ion m/z) and the theoretical mass of the peptide identified. If the resulting value is outside of a normal range (depending on the accuracy of the mass spectrometer used), this constitutes a good indication that something has gone wrong while annotating the data. See details here and in the following open access article.
If the distribution of all the ‘delta m/z’ values from the whole experiment (MS run) is taken into account, it can give a clear indication that something has gone wrong in the experimental set up, or that there has been a mistake in the reporting of the final results at a global level. This chart is available in the ‘Summary charts’ view in PRIDE Inspector.
So what can you do in case you see outliers:
- First decide whether there is a consistent pattern, for instance if the same charge state yields the same consistent mass delta values. Usually charge state misassignments result in much bigger 'delta m/z' values in the range of the (multiples of the) whole peptide masses while modification problems result in much lower 'delta m/z' values.
- In case it looks like a protein modification annotation related problem, check whether you have left out some modifications or that the modification that you have picked during the conversion process in PRIDE Converter 2 are the correct ones (for instance, not use upper more generic terms in the ontology, since they do not have a mass delta). Also it is possible that hey are not represented in the PSI-MOD ontology at all. In that case, those terms need to be added to the PSI-MOD ontology. Contact pride support (pride-support@ebi.ac.uk) for highlighting this issue.
6. Recommendations for popular proteomics software tools and search engines - What fits my custom pipeline?
What kind of Scaffold files are recommended for a PX Submission?
Scaffold 4.0 can export mzIdentML 1.1 files and we recommend uploading those files with the accompanying mgf peak list files that are referenced in them. This way an mzIdentML based Complete Submission can be done.
For older Scaffold versions we recommend exporting the binary .sf3 result files as ProtXML files and use them for a Partial Submission as "search" files.
What kind of MaxQuant files are recommended for a PX Submission?
MaxQuant output is not supported by PRIDE Converter 2 and at present there is no exporter to any of the supported formats. A Partial ProteomeXchange Submission is then recommended where MaxQuant output files should be uploaded alongside with the 'RAW' files.
If you are using MaxQuant (version 1.3.0.5) there is a txt folder generated and by default you can just zip this text folder and upload as search/identification file.
If this is complicated we would recommend uploading the following particular text output files:
peptides.txt
modifiedPeptides.txt
proteinGroups.txt
msms.txt
parameters.txt
and your Experimental Design Template file saved as a tab separated file.
It is also recommended to upload the corresponding folder, usually entitled 'Andromeda', containing the .apl peak list files as a compressed zip file and annotate it as a PEAK file as it helps with PSM recovery.
What kind of Proteome Discoverer files are recommended for a PX Submission?
1. For a Partial Submission please export human readable pep.xml files out of the .msf files with Proteome Discoverer and upload the pepXML files and msf files as 'SEARCH' files for a Partial Submission, please see www.ebi.ac.uk/pride/help/archive/faq#partial-PX.
2. In PD 2.2 the mzIdentML files are unfortunately not suitable for complete submission, yet. The PD team is working in solving this problem, but we are not aware of the current status. You would need to contact Thermo support to get more details.
3. If you are aiming for a Complete Submission please try to convert the binary .msf files into mzIdentML files with the ProCon tool (Bochum University) which is still under testing. Please see documentation and please note that ProCon only supports PD 1.x files.
What kind of ProteinPilot files are recommended for a PX Submission?
From version 5.0 ProteinPilot can export mzIdentML files, but these are version 1.2 and currently a Complete Submission can only accept 1.1 mzid files.
Generally a Partial Submission can be done and we strongly recommend providing human readable files besides the binary group file. Please export the group files into XML files using the following command line feature:
"Command Line Control and Open Results
To support users and third-party software vendors that want to integrate ProteinPilot&trade Software, it is possible to script searches via command line and decrypt the .group file results into clear XML for full access to all the data it contains."
Here is a howto on the conversion process from one of the PX submitters:
1. Create a txt file in Notepad entitled say "group2XML_Example.bat.txt" and save it in the ProteinPilot folder (where the group2xml.exe is located).
2. Rename "group2XML_Example.bat.txt" to "group2XML_Example.bat", giving it a Windows batch file extension.
3. Opened this batch file in Notepad and type in the following command line instructions:
group2XML.exe XML <full path to the .group file to be converterd> <full path to the .xml file the .group file will be converted into>
for instance
group2XML.exe XML "C:\AB SCIEX\ProteinPilot Data\Results\Example.group" "C:\AB SCIEX\ProteinPilot Data\Results\Example.xml"
The command has the following argument structure: group2XML.exe <Type> <Result.group> <Output.file>
where:
- <Type> specifies the type of output
- <Result.group> is a .group file created by ProteinPilot Software
- <Output.file> is the name of the file to be created
4. Save and close the file.
5. Double-click on the file to run the conversion.
7. Recommendations for specific proteomics experiment types - What might fit my bleeding edge research?
I have mass spectrometry imaging data, what should I do?
There is special support with different file type covering MS Imaging datasets but these can only be submitted as Partial submissions.
These are the main specific points to consider for this type of submissions. See all the details in this open access publication in the journal Analytical and Bioanalytical Chemistry (Springer).
(i) Additional file tags have been created: metadata information about the images (labeled as ‘MS_IMAGE_DATA’) and an optical image (labeled as ‘OPTICAL’).
(ii) It is mandatory to provide the MS raw data (called ‘RAW’).
- It is recommended to submit MS imaging data in the imzML format as it offers the most flexible options for viewing, but proprietary data formats are also accepted.
- There is the possibility to submit two different mass spectral related files for one dataset, as required for several MS imaging data formats (e.g. imzML and Analyze). The mass spectral data file (*.ibd for imzML or *.img file in Analyze format) must be labeled as ‘RAW’. The file that contains metadata (such as pixel dimensions and additional information) must be labeled as ‘MS_IMAGE_DATA’ (e.g. *.imzml file for imzML or *.hdr file for Analyze).
- If an ‘ibd file (imzML format) is submitted as ‘RAW’ an ‘MS_IMAGE_DATA’ (*.imzml) is mandatory.
- However, in the case of ‘RAW’ proprietary formats that only consist of one file, a ‘MS-IMAGE_DATA’ file is not required.
(iii) In addition, PRIDE requires a mandatory ‘SEARCH’ file for Partial submissions, which corresponds to the processed results. There is currently no strict definition for the format of this mandatory file, but it should contain a list of m/z values, names of (tentatively) identified compounds and additional information that were used to the generate MS images in the published work.
(iv) It is also supported the inclusion of an optical image (‘OPTICAL’) of the measured sample, which can allow validation and/or interpretation. The ‘OPTICAL’ file could contain a photograph of the imaged sample or an adjacent section that shows comparable spatial features. Native samples, classical histological techniques (H&E, toluidine) or immunohistochemistry staining (antibody staining) can be provided for this purpose.
I have multi-omics data uploaded to different databases, what should I do?
The same samples can be investigated with multiple 'omics' technologies, for instance DNA, RNA sequencing, mass spectrometry, NMR, etc. This way for instance transcriptomics, proteomics, and metabolomics datasets are combined to support one study. In case of these 'multi-omics', 'cross-omics' datasets the different datasets are deposited into different repositories.
Database identifiers, accessions of other 'omics' datasets (for instance transcriptomics data uploaded to ArrayExpress, metabolomics data uploaded to Metabolights), URLs can be provided in the optional 'Links to other 'Omics' dataset section of the PX Submission Tool's 'Additional dataset details' window, see screenshot below.

How to handle newly discovered, unreported modifications?
PRIDE supports annotation of protein modifications using both UNIMOD and PSI-MOD.
Most of the mzidentML exporters use UNIMOD.
We recommend using the PSI-MOD ontology to report protein modifications in PRIDE XML files as result files for Complete PX Submissions. In case the protein modifications you were using or have discovered throughout your study have not been reported in the ontology we recommend adding the modification first to the ontology. For that, we need the proposed name, delta mass and/or a reference or web URL (if it is a commercial product).
8. Related journal guidelines - What are the known requirements of different proteomics journals?
Does my PX dataset fulfil the requirements of the journal Molecular and Cellular Proteomics?
The authoritative and up to date source for the precise MCP requirements is the journal website, the following information is given for convenience:
Data deposition: The journal Molecular and Cellular Proteomics recommends that all the MS data associated to a manuscript is submitted to a public data repository such as PRIDE/ProteomeXchange. All types of PX submissions provide stable data availability.
Annotated spectra: MCP requires researchers to provide annotated spectra in several cases such as those proteins identified by one or two peptides, or for all peptide identifications containing post-translational modifications (PTMs). In the case of PX submissions:
- Complete submissions meet this requirement. It is possible to see annotated spectra in PRIDE Inspector.
- In the case of Partial submissions, this requirement is met if a free spectral viewer is available for the search engine output files. For instance, for Scaffold output files, there is a free viewer available. There are other cases when this is possible.
Does my PX dataset fulfil the requirements of the journal Proteomics?
The authoritative and up to date source for the precise Proteomics requirements is the journal website, the following information is given for convenience:
The journal Proteomics requires data availability in the case of the "Dataset Briefs" articles and recommend it for the other article types. All PX submissions provide stable data availability to fulfil this requirement.
9. Troubleshooting
PX Submission Tool does not launch
- Ensure Java is installed on your computer: http://www.java.com
- Download the latest version of the PX Submission Tool: http://www.proteomexchange.org/submission
- Extract the PX Submission Tool to a directory.
- From the tool's directory, double-click the jar file, e.g. px-submission-tool-2.0.1.jar
- If 4.) doesn't work, open a command line to the tool's directory and run the command: java -jar <px-submission-tool-X.Y.Z.jar>, e.g. java -jar px-submission-tool-2.0.1.jar
- If this still doesn't work, please contact PRIDE directly: pride-support@ebi.ac.uk and attach the 'px_submission.log' file found in the 'log' sub-directory.
PX Submission tool launches, but I cannot login / make a submission
Are you connected to the Internet via a proxy? If so, with a plain text editor open the 'config.props' file in the 'config' sub-directory and make changes by following the stated instructions within the file.
How to switch the PX Submission Tool from Aspera to FTP?
Should there be problems with the Aspera upload submitters can switch to the slower FTP file transfer protocol by changing the ‘px.upload.protocol = aspera’ line to ‘px.upload.protocol = ftp’ in the plain config.props text file located in the ‘config’ subdirectory in the PX Submission Tool’s working directory.
PRIDE Converter 2 Tool does not launch or cannot convert my files
- Ensure your opearting system is 64 bit, and you have 64 bit Java installed on your computer: http://www.java.com
- Download the latest version of the PRIDE Converter 2 Tool: https://github.com/PRIDE-Toolsuite/pride-converter-2
- Extract the PRIDE Converter 2 Tool to a directory.
- Increase the allocated memory for the tool. With a plain text editor open the 'converter.properties' file then amend the line:
jvm.args=-Xms64M -Xmx1024M
to a higher allowance, e.g.:
jvm.args=-Xms128M -Xmx3G
- From the tool's main directory, double-click the jar file, e.g. pride-converter-2.0-SNAPSHOT.jar
- If that doesn't work, open a command line to the tool's directory and run the command: java -jar <pride-converter-X.Y-SNAPSHOT.jar>, e.g. java -jar pride-converter-2.0-SNAPSHOT.jar
PRIDE Converter 2 Tool still has problems converting my files
Please contact PRIDE directly: pride-support@ebi.ac.uk. It might be the case that your files are simply incompatible with the PRIDE Converter 2 tool.
How do I update my PRIDE account information?
Log into the PRIDE website with your user account details:
www.ebi.ac.uk/pride/archive/login
And then click on 'Edit profile' to change your details and/or password.
PRIDE Inspector Java Web Start does not launch
Due to Oracle's recent Java update, you may experience security related problems with Java Web Start. Please add "http://www.ebi.ac.uk" to the URLs listed in the Security tab of your Java Control Panel.
PRIDE Inspector Tool does not launch
- Ensure Java is installed on your computer: http://www.java.com
- Download the latest version of the PRIDE Inspector Tool: https://github.com/PRIDE-Toolsuite/pride-inspector
- Extract the PRIDE Inspector Tool to a directory.
- From the tool's directory, double-click the jar file, e.g. pride-inspector-2.5.0.jar
- If 4.) doesn't work, open a command line to the tool's directory and run the command: java -jar <pride-inspector-X.Y.Z.jar>, e.g. java -jar pride-inspector-2.5.0.jar
- If this still doesn't work, please contact PRIDE directly: pride-support@ebi.ac.uk and attach the 'pride_inspector.log' file found in the 'log' sub-directory.
Aspera does not connect
Ensure that the following port allows outbound traffic on your router, firewall, or network: TCP 33001.
Aspera connects, but transfers 0% of my files
Ensure that the following port allows outbound traffic on your router, firewall, or network: UDP 33001.
I cannot to log-in with the PX Submission tool
Please confirm that you can log into the main PRIDE website with your account:
http://www.ebi.ac.uk/pride/archive/login
Perhaps you can try changing your password, and then trying again.
All my Java applications don't launch! I'm on a Mac...
You may need to run your Java application by: <right-click> then "open" the .jar file, or update your GateKeeper security settings to allow applications downloaded from 'anywhere' to be run. See the Apple Support website for more details.
What to do if you can't download or save files
This article describes steps to take if you are unable to download or save files using Firefox.
Firefox includes a download protection feature to protect you from malicious or potentially harmful file downloads. If Firefox has blocked an unsafe download, you will see a warning message about the file in the Downloads panel, along with options for handling the pending download. You can open the Downloads panel to view completed and pending downloads by clicking on the Downloads button (the down arrow on your toolbar). See the articles Where to find and manage downloaded files in Firefox and How does built-in Phishing and Malware Protection work? for more information.
Clearing the download history can fix some problems with downloading files:
- Click the Downloads button
 , and then click Show all downloads. The Downloads window will open.
, and then click Show all downloads. The Downloads window will open. - In the Downloads window, click Clear Downloads.
- Close the Downloads window.
Firefox may not be able to download files if there is a problem with the folder in which downloaded files are saved:
- Click the menu button and select Options.Preferences.
- Select the General panel.
- Go to the Downloads sectionFind the Downloads section under Files and Applications.
- Click the BrowseChoosebutton next to the Save files to entry.
- Choose a different download folder for saving files.
- Close the about:preferences page. Any changes you've made will automatically be saved.
If you have tried the above suggestions, you can restore the default Firefox download folder settings:
- Type about:config in the address bar and press EnterReturn.
A warning page may appear. Click I accept the risk!Accept the Risk and Continue to continue to the about:config page. - In the Search field, enter browser.download.
- If any of the following settings have a status of modifiedare in bold text, reset their values. To reset a value, right-clickhold down the Ctrl key while you click the setting and select Reset from the context menuclick the Delete or Reset button, depending on the preference:
- browser.download.dir
- browser.download.downloadDir
- browser.download.folderList
- browser.download.lastDir
- browser.download.useDownloadDir
- If desired, you can revert back to your preferred download folder settings in your Firefox OptionsPreferencesGeneral panel, in the Downloads section under Files and Applications.
If you receive the error <filename> could not be saved, because an unknown error occurred. , your problem might be caused by an interaction with the Safari browser and your operating system.
To fix the problem, open Safari's Preferences and change the Save downloaded files to: setting to a valid folder (such as your Desktop). Then restart Firefox.
If you receive the error <filename> could not be saved, because you cannot change the contents of that folder. Change the folder properties and try again, or try saving in a different location., your problem might be caused by corrupt plist files.
To fix this problem, go to your home directory and delete this preference file:
- ~/Library/Preferences/com.apple.internetconfig.plist
If downloading certain file types does not work, check to make sure Firefox is not set to handle those file types differently from others. See Change what Firefox does when you click on or download a file for instructions on how to view and change how different file types are handled.
Reset download actions for all file types
To reset how all file types are handled by Firefox back to default:
Open your profile folder:
- Click the menu button , click Help and select Troubleshooting Information.From the Help menu, select Troubleshooting Information. The Troubleshooting Information tab will open.
- Under the Application Basics section next to Profile FolderDirectory, click Open FolderShow in FinderOpen Directory. A window will open that contains your profile folder.Your profile folder will open.
- Click the menu button
- Click the Firefox menu and select Exit.Click the Firefox menu at the top of the screen and select Quit Firefox.Click the Firefox menu and select Quit.
- Delete or rename the mimeTypes.rdf file (for example, rename it mimeTypes.rdf.oldhandlers.json file (for example, rename it handlers.json.old ).
- Restart Firefox.
Internet security software, including firewalls, antivirus programs, anti-spyware programs, and others can block certain file downloads. Check the settings in your security software to see if there is an setting that may be blocking downloads.
To diagnose whether Internet security software is causing problems, you can try temporarily disabling it, seeing if downloads work, and then re-enabling the software.
Downloading an executable file (e.g., an .exe or .msi file) may fail, with the Downloads window showing Canceled under the file name.
This happens because Firefox honors your Windows security settings for downloading applications and other potentially unsafe files from the Internet. To resolve this problem, use the solution given below.
Reset system Internet Security settings
You can reset your system Internet security settings in Internet Explorer. See How to reset Internet Explorer settings at Microsoft Support for instructions.
You may be able to diagnose your download problem by following the steps given in the Troubleshoot and diagnose Firefox problems article.
Based on information from Unable to save or download files (mozillaZine KB)
Share this article: http://mzl.la/1BAQCEh
Recent attempts to download the TrueCrypt files here, using Chrome or Firefox (Mozilla uses Google's technology), have been generating false-positive malware infection warnings. They must be false-positives because no change has been made to the files since this page was put up nearly a year ago (May 29th, 2014) and many people have confirmed that the downloaded binaries have not changed and that their cryptographic hashes still match.
Also, the well-known and respected “VirusTotal” site, which scans files through all virus scanners reports ZERO hits out of 57 separate virus scan tests: VirusTotal scan results.
We have no idea where or why Google got the idea that there was anything wrong with these files. This just appears to be “The Google” doing their best to protect us from ourselves. But that does misfire occasionally. We expect it to fix itself within a day or two.
Although the disappearance of the TrueCrypt site, whose ever-presence the Internet community long ago grew to take for granted, shocked and surprised many, it clearly came as no surprise to the developers who maintained the site and its namesake code for the past ten years. An analysis of the extensive changes made to TrueCrypt's swan song v7.2 release, and to the code's updated v3.1 license, shows that this departure, which was unveiled without preamble, was in fact quite well planned.
For reasons that remain a titillating source of hypothesis, intrigue and paranoia, TrueCrypt's developers chose not to graciously turn their beloved creation over to a wider Internet development community, but rather, as has always been their right granted by TrueCrypt's longstanding license, to attempt to kill it off by creating a dramatically neutered 7.2 version that can only be used to view, but no longer to create new, TrueCrypt volumes.
Then, leveraging the perverse and wrongheaded belief that software whose support was just cancelled renders it immediately untrustworthy, they attempted to foreclose on TrueCrypt's current and continued use by warning the industry that future problems would remain unrepaired. This being said of the latest 7.1a version of the code that has been used by millions, without change, since its release in February of 2012, more than 27 months before. Suddenly, for no disclosed reason, we should no longer trust it?
they still “owned” TrueCrypt, and that it was theirs to kill.
But that's not the way the Internet works. Having created something of such enduring value, which inherently requires significant trust and buy-in, they are rightly unable to now take it back. They might be done with it, but the rest of us are not.
The developers' jealousy is perhaps made more understandable by examining the code they have created. It is truly lovely. It is beautifully constructed. It is amazing work to be deeply proud of. Creating something of TrueCrypt's size and complexity, and holding it together as they did across the span of a decade, is a monumental and truly impressive feat of discipline. So it is entirely understandable when they imply, as quoted below, that they don't trust anyone else to completely understand and maintain their creation as they have. Indeed, it will not be easy. They might look at the coding nightmare atrocity that OpenSSL became over the same span of time and think: “Better to kill off our perfect creation than turn it over to others and have it become that.”
TrueCrypt's creators may well be correct. TrueCrypt may never be as pure and perfect as it is at this moment, today—in the form they created and perfected. Their true final version, 7.1a, may be the pinnacle of this story. So anyone would and should be proud to use and to continue to use this beautiful tool as it is today.
TrueCrypt's formal code audit will continue as planned. Then the code will be forked, the product's license restructured, and it will evolve. The name will be changed because the developers wish to preserve the integrity of the name they have built. They won't allow their name to continue without them. But the world will get some future version, that runs on future operating systems, and future mass storage systems.
There will be continuity . . . as an interesting new chapter of Internet lore is born.
Tweets from the @OpenCryptoAudit project:
- At 5:40am, 29 May 2014
We will be making an announcement later today on the TrueCrypt audit and our work ahead. - 9 hours later at 2:40pm, 29 May 2014
We are continuing forward with formal cryptanalysis of TrueCrypt 7.1 as committed, and hope to deliver a final audit report in a few months. - And eight minutes later at 2:48pm, 29 May 2014
We are considering several scenarios, including potentially supporting a fork under appropriate free license, w/ a fully reproducible build.
So it appears that the unexpected (putting it mildly) disappearance of TrueCrypt.org and the startling disavowal of TrueCrypt's bullet proof security will turn out to be a brief disturbance in the force. We should know much more about a trustworthy TrueCrypt in the late summer of 2014.
| No. The TrueCrypt development team's deliberately alarming and unexpected “goodbye and you'd better stop using TrueCrypt” posting stating that TrueCrypt is suddenly insecure (for no stated reason) appears only to mean that if any problems were to be subsequently found, they would no longer be fixed by the original TrueCrypt developer team . . . much like Windows XP after May of 2014. In other words, we're on our own. But that's okay, since we now know that TrueCrypt is regarded as important enough (see tweets above from the Open Crypto Audit and Linux Foundation projects) to be kept alive by the Internet community as a whole. So, thanks guys . . . we'll take it from here. |
| Note that once TrueCrypt has been independently audited it will be the only mass storage encryption solution to have been audited. This will likely cement TrueCrypt's position as the top, cross-platform, mass storage encryption tool. |
My two blog postings on the day, and the day after, TrueCrypt's self-takedown:
My third and final posting about this page, in order to allow feedback.
The posting generated many interesting comments:
And then the TrueCrypt developers were heard from . . . Steven Barnhart (@stevebarnhart) wrote to an eMail address he had used before and received several replies from “David.” The following snippets were taken from a twitter conversation which then took place between Steven Barnhart (@stevebarnhart) and Matthew Green (@matthew_d_green):
|
| TrueCrypt v7.1a installation packages: | Downloads | |
| • TrueCrypt Setup 7.1a.exe (32/64-bit Windows) | 461,857 | |
| • TrueCrypt 7.1a Mac OS X.dmg | 101,003 | |
| • truecrypt-7.1a-linux-x64.tar.gz | 108,811 | |
| • truecrypt-7.1a-linux-x86.tar.gz | 58,660 | |
| • truecrypt-7.1a-linux-console-x64.tar.gz | 44,960 | |
| • truecrypt-7.1a-linux-console-x86.tar.gz | 33,531 | |
| The TrueCrypt User's Guide for v7.1a: | ||
| • TrueCrypt User Guide.pdf | 243,548 | |
| The TrueCrypt v7.1a source code as a gzipped TAR and a ZIP: | ||
| • TrueCrypt 7.1a Source.tar.gz | 34,993 | |
| • TrueCrypt 7.1a Source.zip | 41,505 | |
(Because caution is never foolish.)
- Many sites attempt to assert the authenticity of the files they offer by posting their cryptographic hash values. But if bad guys were able to maliciously alter the downloaded files, they could probably also alter the displayed hashes. Until we have secure DNS (DNSSEC, which will create a secured Internet-wide reference for many things besides IP addresses) the best we can do is host confirmation hashes somewhere else, under the theory that as unlikely as it is that this primary site was hacked, it's significantly less likely that two unrelated sites were both hacked.
So, for those who double-knot their shoelaces, Taylor Hornby (aka FireXware) of Defuse Security is kindly hosting a page of hash values of every file listed above. And, being the thorough cryptographic code auditor that he is, Taylor first verified the files GRC is offering here against several independent archives:
Additional online TrueCrypt sites and repositories:
- The reconstructed browsable version of the truecrypt.org website. A terrific Canadian web developer, Andrew Y. (who also created the ScriptSafe Chrome browser extension to duplicate the script-disabling of Firefox's NoScript), captured some of the TrueCrypt.org website before it disappeared from the Internet and then reconstructed the missing pieces using the PDF manual. The result is a terrific web-browsable site for TrueCrypt.
- TrueCrypt.ch: A just launched, Swiss-based, possible new home for TrueCrypt. Follow these folks on Twitter: @TrueCryptNext. Given the deliberate continuing licensing encumbrance of the registered TrueCrypt trademark, it seems more likely that the current TrueCrypt code will be forked and subsequently renamed. In other words . . . for legal reasons it appears that what TrueCrypt becomes will not be called “TrueCrypt.”
- github.com/DrWhax/truecrypt-archive: This is a frequently cited and trusted archive maintained by Jurre van Bergen (@DrWhax) and Stefan Sundin. It contains a nearly complete, historical repository of previous TrueCrypt versions, tracking its evolution all the way back to when it was previously named “ScramDisk” (which is when we were first using and working with it).
- github.com/syglug/truecrypt: Another TrueCrypt v7.1 archive, though apparently not the latest. But readily browsable if someone wishes to poke around within the source with their web browser.
- IsTrueCryptAuditedYet.com: This is the home of the TrueCrypt auditing project. As the audit moves into its next phase, digging past the startup and boot loader and into the core crypto, updates will be posted and maintained here.
Graphic designer William Culver spend a bit of time thinking about a logo for whatever TrueCrypt becomes in the future. The theme of an infinity symbol is meant to convey the endless lifetime of this terrific data encryption solution. As is made clear on William's page for this, he's releasing all copyright:

256 x 256 pixels

32 x 32 pixels
Additional Miscellany:
- Amazon uses TrueCrypt when exporting archived data to users. See the first Q&A of the link. TrueCrypt is a perfect solution for this. We have every reason to believe that it is utterly bulletproof and only TrueCrypt provides the universal Windows/Mac/Linux platform neutrality that this application requires.
Gibson Research Corporation is owned and operated by Steve Gibson. The contents of this page are Copyright (c) 2020 Gibson Research Corporation. SpinRite, ShieldsUP, NanoProbe, and any other indicated trademarks are registered trademarks of Gibson Research Corporation, Laguna Hills, CA, USA. GRC's web and customer privacy policy. |  |
| Last Edit: May 12, 2015 at 08:21 (1,955.05 days ago) | Viewed 167 times per day |
What’s New in the reason 9 download Archives?
Screen Shot
System Requirements for Reason 9 download Archives
- First, download the Reason 9 download Archives
-
You can download its setup from given links:

reason 9 download Archives & Software

reason 9 download Archives& Serial Key Download
